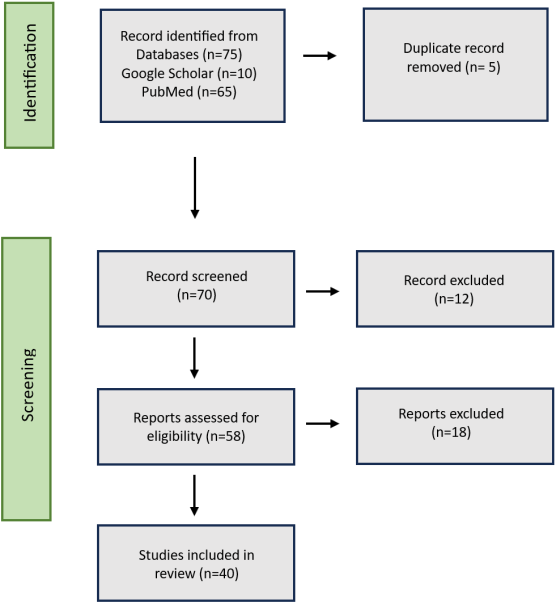
Male infertility in Pakistan exhibits unique genetic patterns due to high consanguinity rates (65%). This systematic review of 38 studies (2015-2025) analyzed 2,041 participants (1,503 infertile men, 538 controls) using whole-exome sequencing (WES). Key findings reveal distinct genetic causes and inheritance patterns specific to this population. Chromosomal abnormalities affected 20.9% of azoospermic men, primarily Klinefelter syndrome (14.7%). Y-chromosome microdeletions occurred in 8% of cases, mostly in the AZFc region (50%). We identified 72 pathogenic variants across 58 genes, with 70.8% being novel to Pakistani populations. Consanguinity drove homozygous inheritance in 72.2% of cases. The most frequently mutated genes included ADAD2 (30% of non-obstructive azoospermia), HFM1 (20%), and DNAH family members (28.7% of motility defects). Variant types comprised frameshift (38.9%), missense (33.3%), nonsense (16.7%), and splicing mutations (11.1%). Significant biochemical markers included the CAT rs7943316 TT genotype (70.9% vs 14% controls) and elevated oxidative stress markers. These findings establish the first comprehensive genetic profile of Pakistani male infertility, demonstrating the profound impact of consanguinity on disease expression. The results emphasize the need for population-specific diagnostic protocols that prioritize DNAH/CFAP genes for motility disorders and ADAD2 for non-obstructive azoospermia. This review provides critical insights for genetic counseling and clinical management in high-consanguinity populations.
| Published in | International Journal of Genetics and Genomics (Volume 13, Issue 4) |
| DOI | 10.11648/j.ijgg.20251304.11 |
| Page(s) | 73-82 |
| Creative Commons |
This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited. |
| Copyright |
Copyright © The Author(s), 2025. Published by Science Publishing Group |
Consanguinity Male Infertility Pakistan, Whole Exome Sequencing (WES), Male Infertility, Consanguineous Marriages, Spermatogenic Failure, Y-chromosome Deletion
Category | Size (N=136) |
|---|---|
Total abnormalities | 28 (20.6%) |
Infertility group | 27/129 (20.9%) |
Miscarriage group | 1/7 (14.2%) |
Numerical abnormalities | 22 (16.2%) |
Klinefelter (47, XXY) | 20 |
Turner (45, X) | - |
Structural Abnormalities | 6 (4.4%) |
Robertsonian translocation | 1 |
Sex reversal (46, XX/46, XY) | 1 |
Category | Number of cases (%) |
|---|---|
Total cases with Yq microdeletions | 12 (5.45%) |
Microdeletions in azoospermia cases | 12/150 (8%) |
AZFa deletion | 1 (8.33%) |
AZFb deletion | 2 (16.67%) |
AZFc deletion | 6 (50%) |
AZFb+c deletion | 2 (16.67%) |
Complete AZF deletion | (8.33%) |
Category | Non-Obstructive Azoospermia (NOA) | Obstructive Azoospermia (OA) |
|---|---|---|
Key Genes | SPATA22, MEIOB, C14orf39, MSH5, HFM1, DND1, KCTD19, ADAD2, ZSWIM7, YTHDC2 | ADGRG2 |
Frequent Mutations | ADAD2 (30%), HFM1 (20%), C14orf39 (10%) | ADGRG2 (100%) |
Mutation Types | Missense (50%), Frameshift (30%), Splicing (10%), Nonsense (10%) | Nonsense (100%) |
Inheritance Pattern | Primarily homozygous (90%); one compound heterozygous case (ADAD2) | X-linked recessive |
Functional Impact | Disrupted meiotic synapsis (C14orf39, MSH5, ZSWIM7), impaired protein interactions (SPATA22-MEIOB, DND1), meiotic arrest (HFM1, KCTD19, ADAD2), RNA metabolism defects (YTHDC2) | Truncated ADGRG2 protein (loss of transmembrane domain) |
Population Frequency | ADAD2 variants in 3 unrelated families; others in single families | Single family reported |
Clinical Diagnosis | Testicular failure (high FSH, small testes) | Normal FSH, palpable vas deferens, post-testicular obstruction |
Histological Findings | Sertoli cell-only, maturation arrest, hypospermatogenesis | Normal spermatogenesis with ductal obstruction |
Treatment Implications | MicroTESE for sperm retrieval (30-50% success) | Surgical reconstruction or sperm retrieval (higher success) |
Gene | Variant | Mutation Type | Zygosity | Phenotypic Impact | Structural Defects | Frequency |
|---|---|---|---|---|---|---|
DNAH17 | c.6308C>T (p.Ala2103Val) c.11803C>T (p.Gln3935*) c.G5408A (p.C1803Y) | Missense Nonsense Missense | Homozygous Homozygous Homozygous | Severe axonemal disorganization | ≥78% abnormal cross-sections, disrupted 9+2 array | 14.3% |
DNAH1 | c.7646_7647insC (p.N2549Qfs*61) c.6212T>G (p.C1789Y) | Frameshift Missense | Homozygous Homozygous | Fibrous sheath defects | Missing central singlets, disorganized FS | 9.5% |
DNAH2 | c.12720G>T (p.W4240C) | Missense | Homozygous | Asthenozoospermia | 80-90% abnormal sperm morphology | 4.8% |
DNAH8 | c.6158_6159insT | Frameshift | Homozygous | MMAF | Divergent flagellar ultrastructure | 4.8% |
DNAH10 | c.9409C>A (p.P3137T) c.12946G>C (p.D4316H) c.8849G>A (p.G2950D) c.11509C>T (p.R3687W) | Missense Missense Missense Missense | Compound Heterozygous | MMAF | Microtubule defects, head abnormalities | 4.8% |
CFAP43 | c.900_901del (p.Arg300Lysfs*22) c.1576_1577del (p.Thr526Serfs*43) | Frameshift Frameshift | Compound Heterozygous Homozygous | MMAF | 82% abnormal cross-sections, absent CPC | 9.5% |
CFAP61 | c.451_452del (p.I151Nfs*4) c.847C>T (p.R283*) | Frameshift Nonsense | Homozygous Homozygous | MMAF | Missing central pairs, absent RS/IDA | 4.8% |
CFAP57 | c.2872C>T (p.R958*) c.2737C>T (p.R913*) | Nonsense Nonsense | Homozygous Homozygous | MMAF | Flagellar assembly defects | 4.8% |
SPAG17 | c.829+1G>T (p.Asp212_Glu276del) c.2120del (p.Leu707*) | Splicing Frameshift | Homozygous Homozygous | CPC defects | Complete CPC absence | 4.8% |
ARMC2 | c.182C>G (p.S61X) | Nonsense | Homozygous | Mitochondrial defects | Vacuolated mitochondria, disrupted flagella | 9.5% |
ARMC3 | c.916+1G>A (p.Glu245_Asp305delfs*16) | Splicing | Homozygous | MMAF | Incomplete C1a, missing doublets 1/9 | 9.5% |
NPHP4 | c.1490C>G (p.P497R) | Missense | Homozygous | 9+0 configuration | Absent central microtubules | 4.8% |
TTC12 | c.1069C>T (p.Arg357Trp) | Missense | Homozygous | Flagellar defects | Ultrastructural abnormalities | 4.8% |
CCDC34 | c.848C>A (p.A283E) | Missense | Homozygous | OAT | Missing outer dynein arms | 4.8% |
AK7 | c.871-4ACA>A | Splicing | Homozygous | MMAF | Disorganized axonemes, abnormal mitochondria | 4.8% |
ADCY10 | c.2902C>T (p.Arg968*) c.4286+1G>T c.436+2T>G | Nonsense Splicing Splicing | Compound Heterozygous Homozygous | Midpiece defects | Misarranged mitochondrial sheaths | 4.8% |
TBC1D25 | c.197A>C (p.Glu50Ala) | Missense | X-linked | Oligozoospermia | Autophagy impairment | 4.8% |
STK33 | c.1235del (p.T412Kfs*14) | Frameshift | Homozygous | MMAF | Complete flagellar disorganization | 4.8% |
ACTL7A | c.149_150del (p.E50Afs*6) | Frameshift | Homozygous | Acrosomal defects | 98.9% acrosomal detachment (founder variant) | 4.8% |
ENO4 | c.293A>G (p.Lys98Arg) | Missense | Homozygous | Sperm head defects | Globular/pyriform heads, reduced size | 4.8% |
AChE | Acetylcholinesterase |
AZF | Azoospermia Factor (regions a, b, c) |
CAT | Catalase |
CFAP | Cilia and Flagella Associated Protein |
CI | Confidence Interval |
CPC | Central Pair Complex |
FS | Fibrous Sheath |
FSH | Follicle-Stimulating Hormone |
GTG-banding | G-banding using Trypsin and Giemsa |
GSH | Glutathione |
HSP90 | Heat Shock Protein 90 |
IL-1β | Interleukin-1 beta |
LH | Luteinizing Hormone |
MMAF | Multiple Morphological Abnormalities of the Flagella |
MTHFR | Methylenetetrahydrofolate Reductase |
NOA | Non-Obstructive Azoospermia |
| [1] | Khayat, A. M., et al., Consanguineous marriage and its association with genetic disorders in Saudi Arabia: a review. Cureus, 2024. 16(2). |
| [2] | Chisholm, J. S. and A. H. Bittles, Consanguinity and the developmental origins of health and disease. J Evol Med, 2015. 3: p. 1-4. |
| [3] | Riaz, H. F., S. Mannan, and S. Malik, Consanguinity and its socio-biological parameters in Rahim yar Khan district, Southern Punjab, Pakistan. Journal of Health, Population and Nutrition, 2016. 35: p. 1-11. |
| [4] | Bittles, A. H. and M. L. Black, Consanguinity, human evolution, and complex diseases. Proceedings of the National Academy of Sciences, 2010. 107(suppl_1): p. 1779-1786. |
| [5] | Oniya, O., et al., A review of the reproductive consequences of consanguinity. European Journal of Obstetrics & Gynecology and Reproductive Biology, 2019. 232: p. 87-96. |
| [6] | Anwar, W. A., M. Khyatti, and K. Hemminki, Consanguinity and genetic diseases in North Africa and immigrants to Europe. The European Journal of Public Health, 2014. 24(suppl_1): p. 57-63. |
| [7] | Bener, A. and R. R. Mohammad, Global distribution of consanguinity and their impact on complex diseases: Genetic disorders from an endogamous population. Egyptian Journal of Medical Human Genetics, 2017. 18(4): p. 315-320. |
| [8] | Demirtas, A., E. C. Akinsal, and O. Ekmekcioglu, Parental consanguinity in infertile males. 2013. |
| [9] | Heidari, F., et al., Prevalence and risk factors of consanguineous marriage. European Journal of General Medicine, 2014. 11(4): p. 248-255. |
| [10] | Lawrenz, B., et al., Ethnical and sociocultural differences causing infertility are poorly understood-insights from the Arabian perspective. Journal of assisted reproduction and genetics, 2019. 36(4): p. 661-665. |
| [11] | Inhorn, M. C., Middle Eastern masculinities in the age of new reproductive technologies: male infertility and stigma in Egypt and Lebanon. Medical anthropology quarterly, 2004. 18(2): p. 162-182. |
| [12] | Minhas, S., et al., European association of urology guidelines on male sexual and reproductive health: 2021 update on male infertility. European urology, 2021. 80(5): p. 603-620. |
| [13] | Ferlin, A., et al., Association of partial AZFc region deletions with spermatogenic impairment and male infertility. Journal of medical genetics, 2005. 42(3): p. 209-213. |
| [14] | Carrell, D., C. De Jonge, and D. Lamb, The genetics of male infertility: a field of study whose time is now. Archives of andrology, 2006. 52(4): p. 269-274. |
| [15] | Krausz, C., Editorial for the special issue on the molecular genetics of male infertility. Human Genetics, 2021. 140(1): p. 1-5. |
| [16] | Nieschlag, E., et al., Male reproductive health and dysfunction. Male reproductive health and dysfunction, 2010. |
| [17] | Waclawska, A. and M. Kurpisz, Key functional genes of spermatogenesis identified by microarray analysis. Systems biology in reproductive medicine, 2012. 58(5): p. 229-235. |
| [18] | Verstovsek, S., et al., Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. New England Journal of Medicine, 2010. 363(12): p. 1117-1127. |
| [19] | Mahmood, A., Y Chromosome Microdeletions in Pakistani Infertile Men. Journal of Islamabad Medical & Dental College, 2017. 6(1): p. 14-21. |
| [20] | Naz, F., et al., Cytogenetic abnormalities associated with reproductive failure in Pakistani population: Experience of a tertiary care hospital. Journal of Pakistan Medical Association, 2024. 74(3): p. 555-555. |
| [21] | Cooper, T. G., et al., World Health Organization reference values for human semen characteristics. Human reproduction update, 2010. 16(3): p. 231-245. |
| [22] | Esteves, S. C., R. Miyaoka, and A. Agarwal, An update on the clinical assessment of the infertile male. Clinics, 2011. 66(4): p. 691-700. |
| [23] | Aziz, N., The importance of semen analysis in the context of azoospermia. Clinics, 2013. 68: p. 35-38. |
| [24] | Esteves, S. C. and A. Agarwai, The azoospermic male: current knowledge and future perspectives. Clinics, 2013. 68: p. 01-04. |
| [25] | Wu, Y., et al., Whole-exome sequencing of consanguineous families with infertile men and women identifies homologous mutations in SPATA22 and MEIOB. Human Reproduction, 2021. 36(10): p. 2793-2804. |
| [26] | Fan, S., et al., Homozygous mutations in C14orf39/SIX6OS1 cause non-obstructive azoospermia and premature ovarian insufficiency in humans. The American Journal of Human Genetics, 2021. 108(2): p. 324-336. |
| [27] | Gong, C., et al., A homozygous loss-of-function mutation in MSH5 abolishes MutSγ axial loading and causes meiotic arrest in NOA-affected individuals. International Journal of Molecular Sciences, 2022. 23(12): p. 6522. |
| [28] | Xie, X., et al., Biallelic HFM1 variants cause non-obstructive azoospermia with meiotic arrest in humans by impairing crossover formation to varying degrees. Human Reproduction, 2022. 37(7): p. 1664-1677. |
| [29] | Xie, X., et al., A homozygous missense variant in DND1 causes non-obstructive azoospermia in humans. Frontiers in Genetics, 2022. 13: p. 1017302. |
| [30] | Zhang, H. and Q. Zhang, Loss-of-function variants in KCTD19 cause non-obstructive azoospermia in humans. |
| [31] | Shi, B., et al., Biallelic mutations in RNA-binding protein ADAD2 cause spermiogenic failure and non-obstructive azoospermia in humans. Human Reproduction Open, 2023. 2023(3): p. hoad022. |
| [32] | Hussain, S., et al., A novel homozygous variant in homologous recombination repair gene ZSWIM7 causes azoospermia in males and primary ovarian insufficiency in females. European Journal of Medical Genetics, 2022. 65(11): p. 104629. |
| [33] | Tian, S., et al., A homozygous missense variant in YTHDC2 induces azoospermia in two siblings. Molecular Genetics and Genomics, 2024. 299(1): p. 84. |
| [34] | Khan, M. J., et al., X-linked ADGRG2 mutation and obstructive azoospermia in a large Pakistani family. Scientific Reports, 2018. 8(1): p. 16280. |
| [35] | Zhang, B., et al., Novel loss‐of‐function variants in DNAH17 cause multiple morphological abnormalities of the sperm flagella in humans and mice. Clinical Genetics, 2021. 99(1): p. 176-186. |
| [36] | Khan, R., et al., Novel loss-of-function mutations in DNAH1 displayed different phenotypic spectrum in humans and mice. Frontiers in Endocrinology, 2021. 12: p. 765639. |
| [37] | Hwang, J. Y., et al., Genetic defects in DNAH2 underlie male infertility with multiple morphological abnormalities of the sperm flagella in humans and mice. Frontiers in Cell and Developmental Biology, 2021. 9: p. 662903. |
| [38] | Dil, S., et al., A novel homozygous frameshift variant in DNAH8 causes multiple morphological abnormalities of the sperm flagella in a consanguineous Pakistani family. Asian Journal of Andrology, 2023. 25(3): p. 350-355. |
| [39] | Shoaib, M., et al., Novel bi-allelic variants in DNAH10 lead to multiple morphological abnormalities of sperm flagella and male infertility. Asian Journal of Andrology, 2025: p. 10.4103. |
| [40] | Khan, I., et al., Novel biallelic loss-of-function mutations in CFAP43 cause multiple morphological abnormalities of the sperm flagellum in Pakistani families. Asian Journal of Andrology, 2021. 23(6): p. 627-632. |
| [41] | Ma, A., et al., Biallelic variants in CFAP61 cause multiple morphological abnormalities of the flagella and male infertility. Frontiers in Cell and Developmental Biology, 2022. 9: p. 803818. |
| [42] | Ma, A., et al., Loss-of-function mutations in CFAP57 cause multiple morphological abnormalities of the flagella in humans and mice. JCI insight, 2023. 8(3): p. e166869. |
| [43] | Liu, T., et al., Novel homozygous SPAG17 variants cause human male infertility through multiple morphological abnormalities of spermatozoal flagella related to axonemal microtubule doublets. Asian Journal of Andrology, 2025. 27(2): p. 245-253. |
| [44] | Khan, I., et al., A novel stop-gain mutation in ARMC2 is associated with multiple morphological abnormalities of the sperm flagella. Reproductive BioMedicine Online, 2021. 43(5): p. 913-919. |
| [45] | Rahim, F., et al., A homozygous ARMC3 splicing variant causes asthenozoospermia and flagellar disorganization in a consanguineous family. Clinical Genetics, 2024. 106(4): p. 437-447. |
| [46] | Ali, A., et al., A novel NPHP4 homozygous missense variant identified in infertile brothers with multiple morphological abnormalities of the sperm flagella. Journal of Assisted Reproduction and Genetics, 2024. 41(1): p. 109-120. |
| [47] | Ali, I., et al., A novel homozygous missense TTC12 variant identified in an infertile Pakistani man with severe oligoasthenoteratozoospermia and primary ciliary dyskinesia. Molecular Genetics and Genomics, 2024. 299(1): p. 69. |
| [48] | Ahmad, N., et al., A novel missense mutation of CCDC34 causes male infertility with oligoasthenoteratozoospermia in a consanguineous Pakistani family. Asian Journal of Andrology, 2024. 26(6): p. 605-609. |
| [49] | Hussain, A., et al., A novel homozygous splicing mutation in AK7 causes multiple morphological abnormalities of sperm flagella in patients from consanguineous Pakistani families. Asian Journal of Andrology, 2025. 27(2): p. 189-195. |
| [50] | Zeb, A., et al., Novel biallelic ADCY10 variants cause asthenozoospermia with excessive residual cytoplasm and hydronephrosis in humans. Reproductive BioMedicine Online, 2025. 50(3): p. 104481. |
| [51] | Nawaz, S., et al., First evidence of involvement of TBC1D25 in causing human male infertility. European Journal of Medical Genetics, 2021. 64(2): p. 104142. |
| [52] | Ma, H., et al., Novel frameshift mutation in STK33 is associated with asthenozoospermia and multiple morphological abnormalities of the flagella. Human Molecular Genetics, 2021. 30(21): p. 1977-1984. |
| [53] | Zhou, J., et al., A recessive ACTL7A founder variant leads to male infertility due to acrosome detachment in Pakistani Pashtuns. Clinical Genetics, 2023. 104(5): p. 564-570. |
| [54] | Nawaz, S., et al., A variant in sperm‐specific glycolytic enzyme enolase 4 (ENO4) causes human male infertility. The Journal of Gene Medicine, 2024. 26(1): p. e3583. |
| [55] | Ullah, N., et al., MTHFR polymorphisms as risk for male infertility in Pakistan and its comparison with socioeconomic status in the world. Personalized Medicine, 2019. 16(1): p. 35-49. |
| [56] | Irfan, M., et al., Association of the MTHFR C677T (rs1801133) polymorphism with idiopathic male infertility in a local Pakistani population. Balkan Journal of Medical Genetics: BJMG, 2016. 19(1): p. 51. |
| [57] | Soda, M., et al., Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature, 2007. 448(7153): p. 561-566. |
| [58] | Sadia, K., et al., Antioxidant enzymes and association of CAT SNP‐21 A/T (rs7943316) with male infertility. Molecular Reproduction and Development, 2021. 88(9): p. 598-604. |
| [59] | Sadia, K., et al., Acetylcholinesterase, pro-inflammatory cytokines, and association of ACHE SNP rs 17228602 with male infertility. PloS one, 2023. 18(4): p. e0282579. |
| [60] | Amjad, S., et al., Correlation of serum kisspeptin and leptin with non-obstructive azoospermia: A cross-sectional study in a subset of Karachi population. Pakistan Journal of Medical Sciences, 2024. 40(3Part-II): p. 410. |
| [61] | Bello, J. H., et al., Dysregulation of mitochondrial sirtuin genes is associated with human male infertility. Andrologia, 2022. 54(1): p. e14274. |
| [62] | Ashraf, M., et al., Association of trinucleotide repeat polymorphisms CAG and GGC in exon 1 of the Androgen Receptor gene with male infertility: a cross-sectional study. Turkish journal of medical sciences, 2022. 52(6): p. 1793-1801. |
APA Style
Abbas, M., Ahmad, N., Faraz, A., Younas, M., Zain-Ul-Abideen, et al. (2025). A Systematic Review of Genetic Causes of Male Infertility Diagnosed by Whole-Exome Sequencing in the Pakistani Population: Updates from 2015 to June 2025. International Journal of Genetics and Genomics, 13(4), 73-82. https://doi.org/10.11648/j.ijgg.20251304.11
ACS Style
Abbas, M.; Ahmad, N.; Faraz, A.; Younas, M.; Zain-Ul-Abideen, et al. A Systematic Review of Genetic Causes of Male Infertility Diagnosed by Whole-Exome Sequencing in the Pakistani Population: Updates from 2015 to June 2025. Int. J. Genet. Genomics 2025, 13(4), 73-82. doi: 10.11648/j.ijgg.20251304.11
AMA Style
Abbas M, Ahmad N, Faraz A, Younas M, Zain-Ul-Abideen, et al. A Systematic Review of Genetic Causes of Male Infertility Diagnosed by Whole-Exome Sequencing in the Pakistani Population: Updates from 2015 to June 2025. Int J Genet Genomics. 2025;13(4):73-82. doi: 10.11648/j.ijgg.20251304.11
@article{10.11648/j.ijgg.20251304.11,
author = {Musavir Abbas and Nisar Ahmad and Ahmad Faraz and Manahil Younas and Zain-Ul-Abideen and Wasim Shah},
title = {A Systematic Review of Genetic Causes of Male Infertility Diagnosed by Whole-Exome Sequencing in the Pakistani Population: Updates from 2015 to June 2025
},
journal = {International Journal of Genetics and Genomics},
volume = {13},
number = {4},
pages = {73-82},
doi = {10.11648/j.ijgg.20251304.11},
url = {https://doi.org/10.11648/j.ijgg.20251304.11},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.ijgg.20251304.11},
abstract = {Male infertility in Pakistan exhibits unique genetic patterns due to high consanguinity rates (65%). This systematic review of 38 studies (2015-2025) analyzed 2,041 participants (1,503 infertile men, 538 controls) using whole-exome sequencing (WES). Key findings reveal distinct genetic causes and inheritance patterns specific to this population. Chromosomal abnormalities affected 20.9% of azoospermic men, primarily Klinefelter syndrome (14.7%). Y-chromosome microdeletions occurred in 8% of cases, mostly in the AZFc region (50%). We identified 72 pathogenic variants across 58 genes, with 70.8% being novel to Pakistani populations. Consanguinity drove homozygous inheritance in 72.2% of cases. The most frequently mutated genes included ADAD2 (30% of non-obstructive azoospermia), HFM1 (20%), and DNAH family members (28.7% of motility defects). Variant types comprised frameshift (38.9%), missense (33.3%), nonsense (16.7%), and splicing mutations (11.1%). Significant biochemical markers included the CAT rs7943316 TT genotype (70.9% vs 14% controls) and elevated oxidative stress markers. These findings establish the first comprehensive genetic profile of Pakistani male infertility, demonstrating the profound impact of consanguinity on disease expression. The results emphasize the need for population-specific diagnostic protocols that prioritize DNAH/CFAP genes for motility disorders and ADAD2 for non-obstructive azoospermia. This review provides critical insights for genetic counseling and clinical management in high-consanguinity populations.
},
year = {2025}
}
TY - JOUR T1 - A Systematic Review of Genetic Causes of Male Infertility Diagnosed by Whole-Exome Sequencing in the Pakistani Population: Updates from 2015 to June 2025 AU - Musavir Abbas AU - Nisar Ahmad AU - Ahmad Faraz AU - Manahil Younas AU - Zain-Ul-Abideen AU - Wasim Shah Y1 - 2025/10/10 PY - 2025 N1 - https://doi.org/10.11648/j.ijgg.20251304.11 DO - 10.11648/j.ijgg.20251304.11 T2 - International Journal of Genetics and Genomics JF - International Journal of Genetics and Genomics JO - International Journal of Genetics and Genomics SP - 73 EP - 82 PB - Science Publishing Group SN - 2376-7359 UR - https://doi.org/10.11648/j.ijgg.20251304.11 AB - Male infertility in Pakistan exhibits unique genetic patterns due to high consanguinity rates (65%). This systematic review of 38 studies (2015-2025) analyzed 2,041 participants (1,503 infertile men, 538 controls) using whole-exome sequencing (WES). Key findings reveal distinct genetic causes and inheritance patterns specific to this population. Chromosomal abnormalities affected 20.9% of azoospermic men, primarily Klinefelter syndrome (14.7%). Y-chromosome microdeletions occurred in 8% of cases, mostly in the AZFc region (50%). We identified 72 pathogenic variants across 58 genes, with 70.8% being novel to Pakistani populations. Consanguinity drove homozygous inheritance in 72.2% of cases. The most frequently mutated genes included ADAD2 (30% of non-obstructive azoospermia), HFM1 (20%), and DNAH family members (28.7% of motility defects). Variant types comprised frameshift (38.9%), missense (33.3%), nonsense (16.7%), and splicing mutations (11.1%). Significant biochemical markers included the CAT rs7943316 TT genotype (70.9% vs 14% controls) and elevated oxidative stress markers. These findings establish the first comprehensive genetic profile of Pakistani male infertility, demonstrating the profound impact of consanguinity on disease expression. The results emphasize the need for population-specific diagnostic protocols that prioritize DNAH/CFAP genes for motility disorders and ADAD2 for non-obstructive azoospermia. This review provides critical insights for genetic counseling and clinical management in high-consanguinity populations. VL - 13 IS - 4 ER -
Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, China
Institute of Biochemistry and Biotechnology, Pir Mehr Ali Shah Arid Agriculture University, Rawalpindi, Pakistan
Institute of Biochemistry and Biotechnology, Pir Mehr Ali Shah Arid Agriculture University, Rawalpindi, Pakistan
Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, China